La mucoviscidose
La mucoviscidose, c'est quoi ?
La mucoviscidose, littéralement "maladie des mucus visqueux", également nommée fibrose kystique du pancréas (ou en anglais cystic fibrosis, CF), est la maladie génétique grave la plus fréquente dans les populations de type caucasienne (population blanche occidentale).
C'est une maladie héréditaire liée à une anomalie de la protéine CFTR. Elle se caractérise par l’épaississement des sécrétions de plusieurs organes, essentiellement les poumons et le pancréas. Les sécrétions ne s'évacuent pas, ce qui altère le fonctionnement de ces organes.
Elle est le plus souvent diagnostiquée dans les premiers mois de vie ou avant 6 ans. Cependant, il existe des formes à révélation plus tardive.
Elle touche en moyenne un nouveau-né sur 4 000 en France, mais 1 sur 3 000 en Bretagne et 1 sur 2 000 dans le Finistère.
Le dépistage de la mucoviscidose systématiquement effectué à la naissance permet un diagnostic précoce.
En France, le nombre de patients atteints de mucoviscidose, bénéficiant d'une prise en charge par l'Assurance Maladie était de 6 960 en 2017 (Source : Assurance Maladie). On estime qu’une personne sur 25 serait porteuse de cette maladie, soit environ deux millions de porteurs sains.
Les symptômes courants de la mucoviscidose sont une toux persistante, un faible gain de poids, une peau au goût très salé chez les bébés.
L'espérance de vie des enfants malades était de 5 ans dans les années 60, de 15 à 20 ans avant la découverte du gène en 1989. Elle est aujourd'hui de plus de 40 ans. | 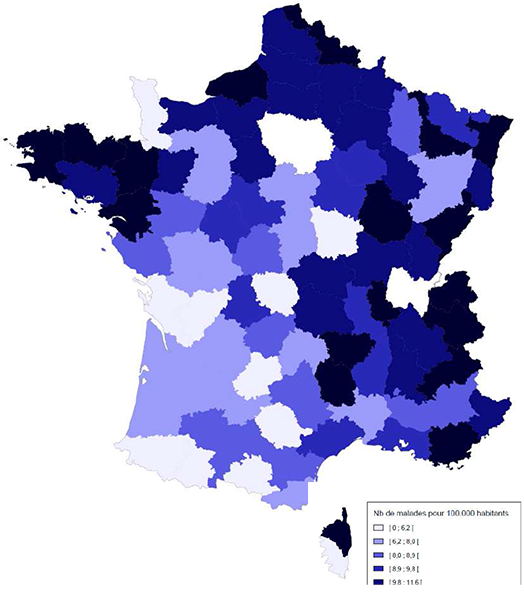 |
Une maladie génétique
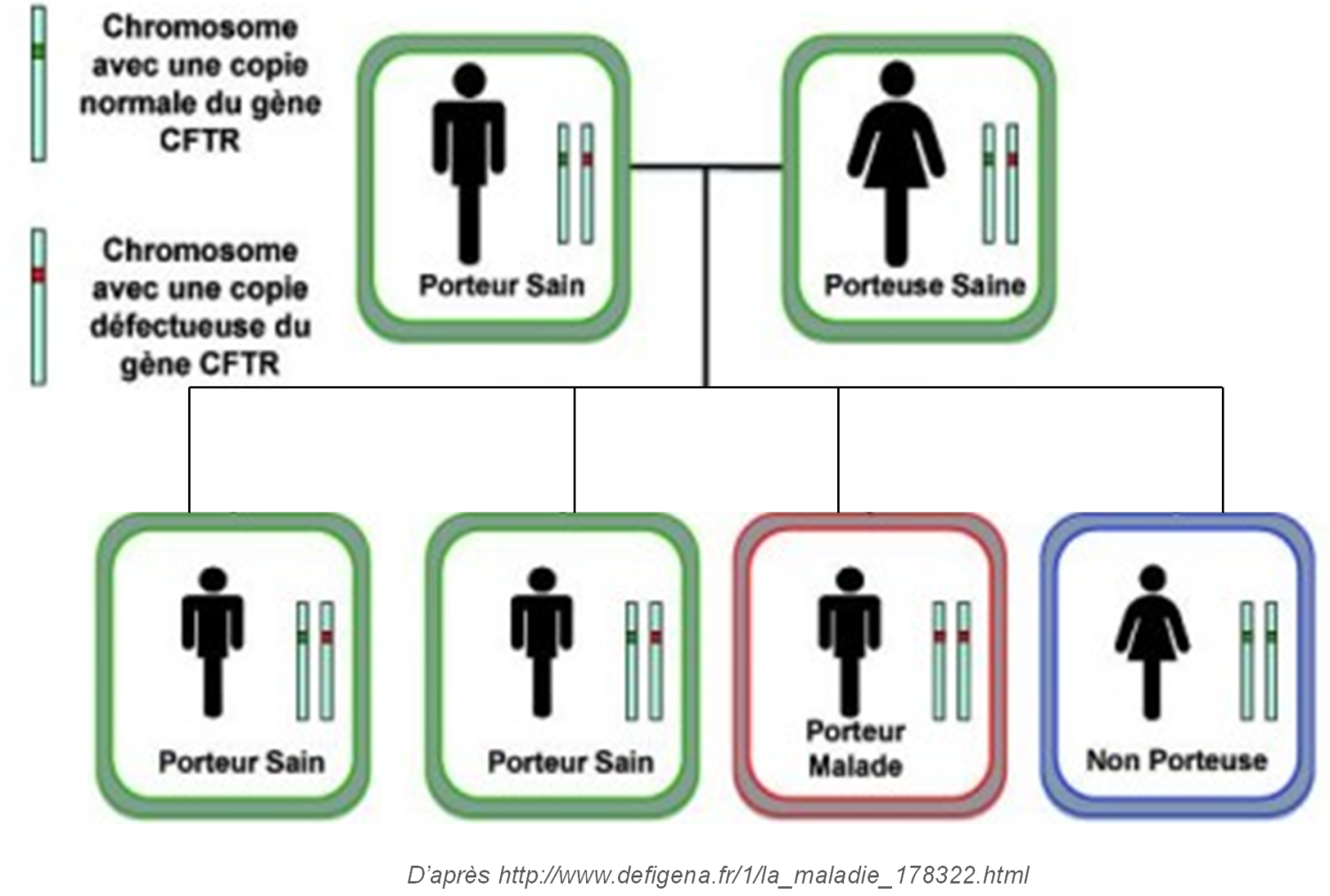 | La mucoviscidose est due à des mutations du gène CFTR (Cystic Fibrosis Transmembrane conductance Regulator).
Elle est transmise selon un mode autosomique récessif, c'est-à-dire qu’elle touche aussi bien les garçons que les filles et que pour être atteint il faut que les deux parents soient porteurs.
Suite à la découverte du gène en 1989, de nombreux laboratoires se sont lancés dans la recherche de mutations du gène CFTR. À ce jour, plus de 2000 anomalies moléculaires du gène CFTR ont été recensées, dont 400 ont été trouvées à Brest. Il existe une mutation principale, communément appelée la mutation F508del. |
Des manifestations cliniques
Le gène CFTR code pour une protéine-canal dont la principale fonction est d’assurer le transport d’ions afin de permettre une hydratation correcte du mucus.
L’absence ou un fonctionnement anormal de cette protéine conduit à une accumulation de mucus visqueux obstruant de nombreux canaux. Différents organes sont alors touchés.
Bien que l’espérance de vie ainsi que la qualité de vie des patients se soient grandement améliorées, la mucoviscidose reste toujours incurable.
La prise en charge thérapeutique des patients repose sur des traitements symptomatiques visant en particulier à améliorer la fonction respiratoire, cause principale de mortalité. | 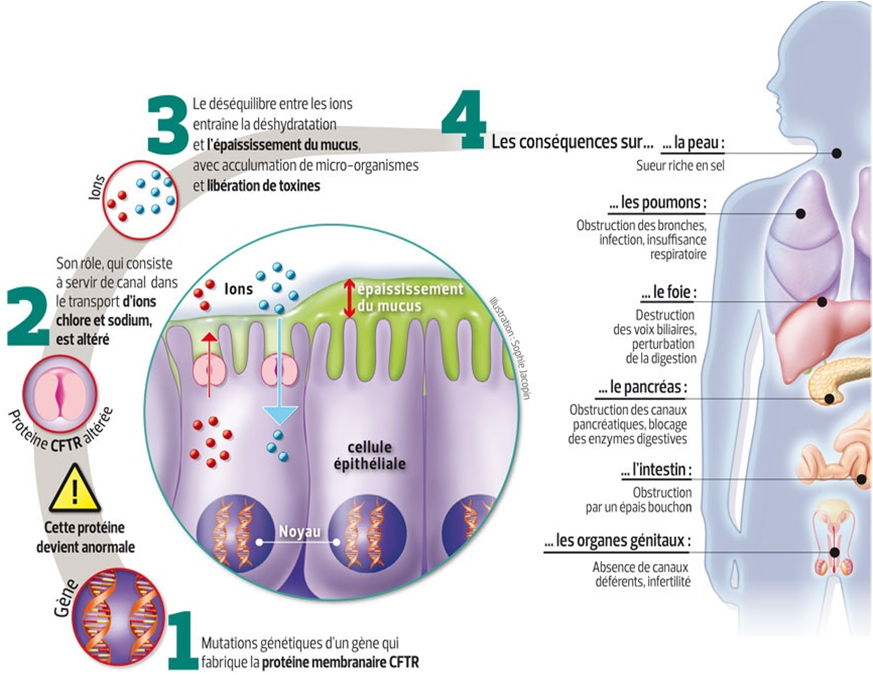 |
Les travaux initiés par le Professeur Claude FÉREC (Unité INSERM U1078) sur la mucoviscidose vont du gène CFTR au malade, en passant par la protéine, et mettent en jeu des travaux de génétique, de protéomique, de signalisation cellulaire mais aussi d’épidémiologie et de transferts de gène.

L'Association Gaétan Saleün est Reconnue d'Utilité Publique, arrêté du 25 mai 1978 et décret du 11 juin 2009.
A ce titre, pour les dons et mécénat des entreprises, elle est habilitée à établir des reçus fiscaux conformément aux articles 200 et 238 du CGI.
L'Association peut également recevoir des legs.